Using Bruker Topas to Process XRD Spectra
Advanced XRD Pattern Processing Using Bruker Topas
The Bruker Topas program is used for advanced processing of xrd scan data, primarily for Rietveld refinement of xrd scans. The program is fairly advanced and requires time to master. There are three manuals that are very helpful in learning how to do things. There are also a number of papers, presentations and tutorials in the Drive D XRD Resources directory on the Rietveld computer
New! We now have Topas version 5 which has several nice improvements; see the Topas manuals for more information.
Here is a summary of the roles that Diffrac.Eva and Topas play in processing of powder diffraction data:
The Eva software is used for basic xrd pattern processing, including search-match and phase identification using the ICDD PDF5+ or COD database files. It has utilities for pattern display and simplified approaches to quantitative analysis, estimation of amorphous content, and compensation for variable unit cell parameters for visual comparision with an expermental powder xrd pattern. One uses Eva to identify a single phase material or all phases present in a multiphase mixture.
The Bruker Topas software is used for advanced powder xrd processing, including Rietveld refinement by whole pattern processing and least squares fitting of peaks defined by structure files to the observed xrd pattern. Assuming that a material has been identified by Eva, the Topas program is used to refine the cell parameters (dimensions and angles), particle size, strain parameters, site occupancies (and atomic positions for single phase refinements), amorphous component, and other parameters. For multiphase mixtures, quantitative analysis is performed using structure files for the known phases and the weight percent of each component in the crystalline fraction is determined by refinement. There are two modes used to run Topas, the GUI interface which is user friendly and can be used to do a lot, and the launch mode, which uses an .inp file and allows for more sophisticated applications. The .inp file contains Topas macros and one needs to be familiar with the Topas Technical Manual to understand the macro language structure. One does not use Topas to identify materials as there is no search match capability.
Processing with Topas includes the identification and fitting of diffraction peaks, assignment of hkl indices and cell refinement, identification of the space group, determination of the crystal structure, and utlimately Rietveld refinement of the structure and aspects of the powder. Corrections are made for the instrumental contribution to the shape of diffraction peaks, strain broadening, differential x-ray microabsorption (multiphase materials), preferred orientation, among others. For materials with unknown structures, the methods of simulated annealing and charge flipping are available to solve the structure. The program has been very successful in solving crystal structures using laboratory powder diffraction data as well as synchrotron data on very large protein crystals. The largest known unit cell (Mo2P2O4) was solved using single crystal data in Topas. Multiple data files can be simultaneously refined so that neutron and x-ray data can be used.
Topas Manuals
You will need to use several resources in order to confidently run the Topas software. There are pdf manuals for the Topas software that you can download, and also several Bruker powerpoint presentations that cover aspects of Topas refinement. I have also provided a basic step-by-step procedure which illustrates how to do a Topas Rietveld refinement.
There are three manuals for Topas version 5:
- The Topas 5 User Manual is a very basic summary of the program that only makes sense after you know how to use the program:
- The windows, menus, and functional parts of the Topas program.
- Various operations used in the program.
- The Topas 5 Tutorial Manual has some examples of how to use the program.
- Profile analysis (peak fitting).
- Unit cell indexing methods.
- Whole pattern decomposition.
- Structure determination by simulated annealing.
- Structure determination by charge flipping.
- Rietveld refinement.
- Quantitative Rietveld analysis (determination of modal abundances in multiphase mixtures).
- Degree of crystallinity determination.
- Isotropic size-strain analysis.
- Using the rigid body editor.
- TOF neutron data.
- Fourier analysis.
There are a number of example data sets that show how Topas is used for processing. These files are located in C:\Topas5\Tutorial on both Bragg and Rietveld computers. These tutorials are discussed in the Topas powerpoint presentations and used in the John Evans Topas web site.
- The Topas 5 Technical Reference Manual has all details of the Topas macro language and built-in commands that are used in launch mode.
Bruker Topas Powerpoint Presentations
Excellent Bruker tutorials on Topas:
- Topas and Rietveld Introduction
- Topas Online training part 1
- Topas Online training part 2
Example Topas Procedures
The following steps show how to do basic Topas Rietveld refinement of a powder xrd pattern.
Startup: Launch the Topas5 program.

Load scan file
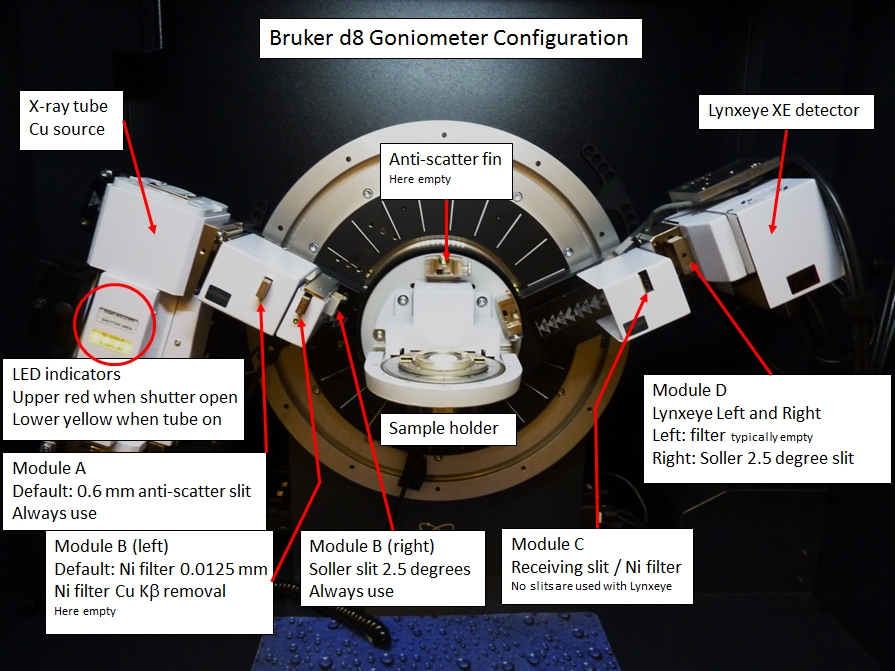
Use File - Load Scan Files to navigate to your scan file and load it. You will load the .raw file, not the .brml file generated by the Bruker software. Very important, note that .raw file has no instrument settings. The range.def file in the Topas5 directory contains default settings for our Bruker d8 Advance. These settings include the goniometer radius (280 mm), the slits typically used (two Soller slits with 2.5 degree acceptance), and the LynxEyeXE detector parameters (3 degree acceptance).
The example file used here is for a quartz sample. If you are processing a file acquired on a different instrument, you will need to edit the instrumental parameters and save your Topas project so that those parameters are used in the refinement.

Default settings
This window shows the default settings for refinement of XRD scans acquired on the WUSTL Bruker d8 Advance. These values are read from the range.def file in the Topas5 folder and assigned by default to all loaded scan data. The settings are as follows, listed top to bottom when the loaded XRD scan file is selected in the tree window:
- Refined vs. fixed parameter values. Any parameter shown in red is a refined parameter and may have the refine keyword adjacent when in grid view. A parameter that is set for refinement may also have the ampersand character @ which indicates it is to be refined. Any parameter that is fixed is either in black text, has the fixed keyword adjacent, or does not have the ampersand character.
- In the window below, only the background is set for refinement. The full axial model and slit parameters are fixed as they are not variable parameters on the instrument.
- Background treatment using Chebychev polynomial of order 3, with 1/x scaling turned on. This fits a 3rd order polynomial to the scan background and includes 1/x to accommodate the increasing air scatter at low 2θ angles. The polynomial order can be changed, but you should avoid using a too-high order as this will potentially fit low intensity peaks rather than background.
- The WUSTL Bruker d8 uses a LynxEyeXE position sensitive detector (Linear PSD), which has a 3 degree 2θ acceptance angle (2Th angular range of LPSD), 0.3 degree FDS angle; these parameters are automatically selected as shown below.
- The Axial Convolution settings should also use Full Axial Model, with the Primary and Secondary Soller slits set to 2.5 degrees.
- Because no monochromator is present, the Lorentz Polarization factor LP factor should be selected but set to zero (see the Topas technical manual for further information regarding LP factor settings).
- If you are refining data collected on another instrument, you will need to set these parameters as necessary, otherwise use the defaults for data collected on the WUSTL Bruker d8.

Emission Profile settings (not refined)
The Emission Profile settings are used to specify the X-ray wavelength of the tube source. The WUSTL Bruker d8 uses a Cu X-ray tube and one can select from the default lamda file which has the wavelength values for Cu Kα1 and Kα2, or other lamda files that contain a more complete listing of the Cu Kα peak bundles. Note that we refine using the Cu Kα peaks in order to fit the doublet seen in the XRD scan.

Background before refinement
This shows the background settings before refinement. After refinement, the fields will be populated with the parameters for the polynomial and 1/x parameters.

Instrument WUSTL Bruker d8
This shows the instrument parameters used for refinement. The WUSTL Bruker d8 has a goniometer radius of 280 mm, LynxEyeXE position sensitive detector with 3 degree acceptance angle, 2.5 degree Soller slits, and we use the Full axial model for instrument correction.

Corrections
Always use Lorentz Polarization correction. For PSD with no monochromator use value of 0 fixed. If processing tutorial files with data acquired on a Bragg Brentano instrument with a monochromator, will need to use value specified in the tutorial.

Miscellaneous
Normally only a subset of the XRD scan range is used for refinement. This reduces the effect of poor counting statistics at higher angles on the refinement. Here you can set the range that is specifically used.

Loading structure data into Topas using the Load STR command
Refinement using Topas requires that you have atomic position data. The easiest procedure is to load this atomic position data into Topas using the Load STR command. Files with a .str extension are in the format required for Topas to read in and use. The latest version of the ICDD PDF5+ database utility on the Rietveld computer allows you to save ICDD card data from the File menu (File - Save PDF card, then select the Topas structure format from the file type pull-down menu). This .str file exported from the ICDD database can then be used to perform refinements.
Topas Structure .str files are located on Drive C in the Topas5 folder, Structure Database subfolder. These .str files have been exported from the PDF5+ program. The .str file is a text file having the Topas script language structure, and contains information such as the space group, cell dimensions, axial angles, cation and anion site xyz values, site occupancy, and other commands used in the Rietveld fitting procedure.
There is a dedicated User Structures folder that each research group should use to store .str files exported from the PDF5+ program. It is recommended that when you export a .str file, you add the phase name at the beginning of the file name and keep the ICDD card number so that you can easily identify the phase by inspection.
You can also use .cif files for Rietveld refinement. The Crystallography Open Database can be used to convert atomic position coordinates to Topas .str structure files. This has been done for Topas5 and the directory COD database for Topas contains COD structure files in the Topas .str format. Finally, if no .str file can be found, or if no ICDD structure data can be used to generate a Topas .str file, then you can use a Crystallography Information Format file with a .cif extension to perform Topas refinements. Note that there is some variation in .cif file formatting and one potential problem is that the cation and anion element symbols may not be read into Topas correctly and you will need to enter them in the sites field.
Options -- Load STR(s) to load a structure file
Right click on the pattern name or select Load STR from the command pane.


After STR loaded
You may wish to compare the .str file with the grid window in Topas to see how the data is loaded into Topas for use in the refinement. The .str file contains data and keywords, but also has variable declaration (including use of the ! flag to inhibit refinement of that parameter), and constraints on the limits that a parameter may obtain during refinement (this can be seen for crystal size and cell axial size values).
Here is the display of quartz structure file parameters after the quart.str structure file was loaded. Again, parameters to be refined are shown in red. The cell axis values are to be refined and will also be assigned to the variables a_quartz and c_quartz. The crystal size has a nominal value of 1000 nm, is to be refined, and can range from a minimum of 32 nm to a maximum that has no limit. Note that if the crystal size L value was refined initially, then the refined value can be fixed; the size parameter can also be turned on and off with the "Use" checkbox.

Display prior to refine
Quartz unit cell parameters are as-loaded, red color indicates refinable quantity. Crystal size is default number and has minimum limit of 32 nm. Note variables used for cell paremeters (a_quartz, c_quartz, etc.) which can be used in Topas macro for reporting or other processing.
Refine using the red play button
The refinement is initiated using the red play button, and typically you are given the choice of accepting or rejecting the refinement results. This is based on a visual inspection of the refined spectrum in case something goes wrong.

Display immediately after refine
Option to accept refinement.

Background after refinement
A 3rd order Chebychev polynomial was used, with 1/X bkg to treat increase in background intensity at low 2θ angles.

Corrections
Most samples do not precisely fill the sample holder so that the sample is on the Roland circle of the goniometer. This excess or deficient sample z-axis location causes the XRD scan to shift to higher or lower 2θ values, respectively. The Sample displacement correction is enabled here and corrects for the offset residual during refinement. This value is in mm, so here the offset is 94 um deficient to the Z=0 reference surface.

Site Values (Quartz example)
Values for site positions x y z for quartz structure. The blue color indicates a special position that must be described using code =1/4 =2/3 etc.

Site Codes (Quartz example)
Codes for site positions x y z for quartz structure. The position for Si4+ is special and set to exactly 2/3 using =2/3 code. Note variable names x1_quartz etc. here !x1_quartz means variable that is fixed (not being refined); removal of the ! mark makes that parameter refinable.

Topas Quantitative Analysis
Quantitative Analysis Example
Quantitative analysis is the determination of the weight percent of phases in a multiphase sample.
The example used is the Bchips sample that is used for EVA search-match demonstrations, it is a mixture of calcite, dolomite, and quartz, with a minor amount of clinochlore (chlorite).
It is very important to understand the following:
- EVA search-match is used to identify the phases in a sample. You must identify all important phases before doing Rietveld refinement.
EVA has a semi-quant option which you should not use, do quant with the Topas program.
- Topas Rietveld refinement is used to perform quantitative analysis or refine the phase information in a sample. It is not used to identify the phases in the sample.
Remember that quantitative analysis can be reduced to the following steps:
- Acquire the XRD scan data using the Bruker Diffrac software.
- Identify the phases in the sample using Bruker EVA.
This includes background subtraction and search-match procedures in order to identify all phases in the pattern. For samples containing minor components you will need to mask out the main peaks and do several iterations of the search-match in order to identify all phases.
- Important: EVA allows you to match using ICDD cards which may or may not have atomic structure coordinates because you are matching peak positions to your XRD pattern. For Topas refinement you must have structure .str files that are based on ICDD cards which have atomic structure positions.
It is also possible to load a .cif file to use for the structure. Note however: .cif files have variable format structure and you will need to confirm that the element symbols have been loaded. If not, you will need to edit the element field in the structure after loaded into Topas.
- Also note that .str files generally do not have particle size refinement turned on, and if this is needed you should use the Microstructure button for each loaded .str to enable Lorentzian refinement of the particle size, and enter constraints for minimum and maximum as needed; if the phase is a minor component, the refinement may iterate to a very small particle size and accommodate the background rather than fitting the phase. In this case a minimum should be enforced.
- Perform quantitative analysis using the Topas Rietveld refinement software coupled with the structure files for all phases identified using EVA. Topas can be used to confirm the correct identification of phases by means of the pattern residual, but is not a search-match program.
To do this, load a scan pattern as outlined previously, set the proper refinement parameters, and load an .str structure file for each phase that has been identified in the sample. You will use the Bruker EVA program to perform search-match and identify those phases, and you must do this before proceeding to using Topas for quantitative analysis.
Bchips multiphase sample - EVA Search Match Results
Here is the EVA program with the Bchips .raw XRD scan loaded, and showing the results of the Search-Match procedure. The sample contains calcite, dolomite, quartz, and a minor contaminant of the chlorite mineral clinochlore. Note that the EVA match results are based on cards that may or may not have atomic coordinates. The clinochlore card initially matched does not have coordinates.

Bchips multiphase sample - Finding a PDF5+ ICDD Card with atomic coordinates
Now using the PDF5+ utility program: We search for the mineral "clinochlore" in the name field of the PDF5+ main window, and select a card that has peaks at the correct position (this is done by inspection of the synthetic pattern and the 2-theta peak listing on the card), and also has the required atomic coordinates as seen here using the Structure button on the left (cards with no atomic positions have no values when this data is viewed). The search results also has a column with a check mark to indicate which cards have atomic coordinates. We confirm this card is appropriate by loading it into EVA using the match by number selection on the left side and compare with the Bchips peak positions as we do for normal Search-Match procedures.


Here is the EVA Search-Match display for the Bchips XRD scan with card matches for calcite, dolomite, quartz. and the clinochlore card matched using SM on the pattern but which does not have atomic coordinates (and is unchecked here), and the clinocholore card found using name search in the PDF5+ program and loaded into EVA using the card number.

Bchips multiphase sample - Exporting PDF5+ ICDD Card with atomic coordinates as a Bruker Topas Structure .str file
Once the correct card is identified in this way, we export it from the PDF5+ program using the Export drop down menu at the upper left of the card display and save it as a Topas *.str structure file.
This structure file should be saved to the User structures directory located on drive C:
C:\Topas5\Structures\User Structures\Your Group folder\Your folder.
Bchips multiphase sample - Topas Rietveld Refinement
Now launch the Topas Rietveld Refinement program. You will need to resize the program to look like this, which includes moving the boundary of the right pane to the left as shown, if you don't resize then you will not be able to see the refinement control button.
Refer back to the Topas instructions for general controls on the refinement setup.
Important Notes regarding refinement and quantitative analysis
- Remember that RED parameters or the @ character denote a quantity that will be refined. If a value is to be fixed then the paramter should be clicked to select "fixed" rather than "refine". The @ symbol is shorthand for "refine".
- While it is possible to refine all parameters simultaneously, the refinement may not converge at all or may not converge to the global minimum Rwp (whole pattern residual). In these cases you may need to refine initial quantiteis, then set them to fixed, and iteratively refine other parameters in order to obtain a final solution.
- Refinement of single phase samples typically includes refinement of cell parameters such as cell dimensions, atomic positions, and site occupancy. In the refinement of multiphase samples for quantitative analysis, parameters such as atomic positions and site occupancy are not refined as there are peak interferences and other complications that result in erroneous values.

Use File menu to load scan data, this will be the .raw scan file saved from a diffraction run. It is a good idea to unzoom the spectrum by right clicking in the scan display area and selecting unzoom. Remember that the refinement is context sensitive and if you are zoomed in on the spectrum, only that region will be refined.

Expand the tree for this scan file to show the controls (Corrections, Background etc.). Here the basic instrument and background settings are displayed.

Here the Corrections are displayed. In general you should refine the Sample Displacement Error because the powder sample is typically higher or lower than the surface of the sample holder. You should not in general refine the zero position as that has been accurately set for the instrument and it is more likely there is a sample displacement error. You may need to refine sample displacement initially and then set to fixed for refinements that are not converging properly.

Here Miscellaneous has been selected in order to restrict the scan range for refinement. In general, you should use the portion of scan range above low angle background curvature which has no diffraction peaks, and set the upper limit to avoid peaks that have comparatively poor counting statistics and are overlapped because these do not contribute to accurate refinement. Here the range has been set to 10 - 54 degrees.

Now load the .str files to be used for refinement. This is done by right clicking on the scan file name (here Bchips.raw with the blue square) and selecting Load STR (do not select Load Structure), or click on the scan file name to highlight and select Load STR from below the tree area. You can load multiple .str files by multiple selection in the dialog window or add them sequentially. Here the .str files for calcite, dolomite, and quartz have been loaded.

The Structures/HKL Phases folder allows you to select which phases to "use" in the refinement. You can also select the "use" checkbox for each phase. Here the calcite structure folder has been opened and the "use" switch, space group number (click on this to see the space group descriptors), the unit cell dimensions which are shown in red for refinable quantities (some structures do not have refinement turned on and should not be refined), and the "Scale" checkbox which will fit the intensity of peaks during refinement and should be turned on.
The Wt% Rietveld value will show the weight percent of the phase here and on the main scan window at far right. The unit cell mass and volume are known quantities and the refined values allow the calculation of weight % of the phase.

Click on the Microstructure button to open this window, which allows you to turn on refinement of Lorentzian particle size calculation and other paramenters (see the Topas Technical Manual for these topics). In general you should use the Lorentzian refinement to correct for particle size effects. If the refined peaks are sharp and narrow compared to the scan data peaks, then particle size refinement is necessary.

Here the Sites folder has been selected. This shows the crystallographically distinct sites, their coordinates in relative unit cell distance, the cation present under the Atom heading along with the valence, the occupancy, and thermal Beq value (see Topas Technical Manual for information on Beq). The use of the =0 and =1/4 is required to avoid roundoff error in unit cell coordinates and these are denoted as "special positions". Again, if a .cif file was used, check to be sure that the Atom entries have been populated; to change them click on the field and select the atom and valence from the popup menu that opens.

Samples that exhibit preferred orientation (acicular or platy materials) may have a peak that is higher than the calculated intensity from the refinement. Fitted intensities are expected to scale according to diffraction from randomly oriented particles. Structure files may include a declared HKL which is a likely reflection to exhibit higher intensity due to preferred orientation. The HKL values are for the peak in question, the "use" checkbox is used to turn the correction on and off, and the fix vs. refine keyword is used to refine or use a previously refined value. You will observe that the declared peak will be fitted independently during refinement with preferred orientation turned on.

With the .str structure files loaded and parameters checked, it is now time to refine the Bchips pattern. Do this by clicking on the red "play" button on the Topas controller at upper right. The "play" button refines to convergence and the "play until end" button refines to completion as determined by the user.

After refinement, a "Refinement Converged" dialog box is opened. The calculated spectrum is shown in red and compared to the blue XRD scan data. If selected for display, the residual is shown below as seen in this display, and can be used to determine the quality of the fit. If the fit is good, click on "Yes" to keep the refined results, but if the refinement did not converge, click on No to identify where the problem might be.
You can select the scan data to be displayed by selecting these items from the View menu.
To get a screen capture of the scan area with the phase wt%, right click in the scan window and click on the entry to copy a bitmap of the scan area to the clipboard.

The weight percent of each phase is displayed at the far right of the scan window. You can use the snipping tool to capture that display for pasting into Powerpoint etc. (there does not seem to be any other place where the phase weight percents are listed).

To view the calculated XRD fit for each phase, hover the mouse pointer over the phase name at the far right of the XRD scan display. This will show the calculated intensity for that phase only.
The following windows show the calculated spectra for calcite, dolomite, and quartz. To return to the main fitted display, click anywhere in the scan window.



Handling Multiple Structure Files in Refinement
It is possible to have several .str files loaded into Topas and use a subset of them. Select the Structures/HKL Phases to see a list of the structures currently loaded into the Topas project. The "use" checkbox allows you to select structures for refinement. Here the crystal size refinement is listed at the top, and the weight percent or amplitude scaling is shown at the bottom. This allows these two sets of parameters to be selected independantly.
It is important to understand that the structure file may not contain the full inventory of elements that are in the phases you are processing. For example, a scan on an ankerite (Mg,Fe,Mn dolomitic carbonate) nominally requires a structure file with the species Ca, Mg, Fe, and Mn listed in the file with their presence declared in the sites display. It is possible however, to refine using structure files for calcite, magnesite, siderite, and rhodochrosite to accomplish this. These structures would be fitted and the weight percent data used to calculate the composition of the natural sample being fitted.

For the Bchips example, it was shown in the EVA instructions how to sequentially perform Search-Match of the XRD scan. First the SM procedure was used to match the calcite, dolomite, and quartz peaks, and then those peaks were masked and the spectrum searched again in order to match the low angle peaks which allows the identification of the chlorite mineral clinochlore. Again, the identification of phases is done using EVA, but the quantitative analysis is done using Topas.
The low angle region of the Topas scan window shows that peaks are present from clinochlore. A .str file can be exported from the PDF5+ database by searching for cards with the phase name "clinochlore", selecting a card which has atomic coordinates, and exporting it as a Topas .str file.

Now the .str structure file for clinochlore has been added to the project, and inspected prior to fitting. It is selected for fitting with the "use" checkbox.

This is now the final refinement for the Bchips example, with calcite, dolomite, quartz, and clinochlore all fitted, and the results shown in the scan window. Note that the scan range for fitting avoids the concave down portion of the scan at low angles. The background model used must not overcorrect the background in the range from 10-18 degrees or the clinochlore concentration will be underestimated. The calculated paricle size for each phase is listed in the upper window, and the phase concentrations are shown at the far right of the main scan window.


Topas Project Files and Processing Additional XRD Scans
A Topas project file contains the current XRD scan data, the structure files, and settings used for refinement. Once you have set up all these items, you should use File Save Project As to save the project. As you work on a project, save it periodically so that if the refinement blows up, you can reload the project and start over. Some divergences result in a situation that cannot be recovered from because multiple parameters have refined to bad values.
If you have additional XRD scans to process with the current project setup, then first save the project with a new name. To load the new XRD scan, select the scan name in the tree window (it has the blue square next to it), right click, and select Replace Scan Data, then navigate to the location of the .raw XRD scan and select it. You can immediately perform the refinement to the extent that all structures and refinement parameters are close enough for the new scan.
Refinement of multiple XRD scans. It is possible to load many XRD scans into the current project. However, you need to select one scan for refinement at a time, this is done by clicking on the colored square next to the scan file name to fill the square, and deselecting all the other scans. Otherwise you fit all scans at once and any variables that are duplicated will cause an error message. It is easier to use one project per scan and use Replace Scan Data to load a new XRD scan file.
Refinement of multiple XRD scans and combined data sets (synchrotron and lab-based diffraction data). The Topas documentation indicates that a refinement can be done on multiple data sets simultaneously. There is no example for this.
End Quantitative Analysis
Bruker Calibration Check Using NIST 1976a Corundum Sample
The Bruker d8 has excellent long-term calibration stability and has a guaranteed specification for both diffraction
peak position and peak intensity using the NIST 1976a Corundum sample mount. It is not necessary to perform daily
checks of calibration as we have done on the older Rigaku DMAX/A, so this step is optional.
If the calibration needs to be checked, use the following setup to perform a run using NIST 1976a.
NIST 1976a Calibration Check:
- Use the Sample Exchange Procedure to change samples and put the NIST 1976a sample in the sample holder.
- Make sure that the following slits are installed on the d8:
- Anti-scatter slit: 0.6 mm.
- No slit or filter in second module.
- Tube-side 2.5 degree Soller slit (normally always in place).
- NIST 1976a sample in holder.
- No slit or filter in third module.
- LynxEyeXE detector: No filter in slot A, 2.5 degree Soller slit in slot B.
Bruker Emergency Procedures
About the Bruker system protection sensors
The Bruker has an extensive set of sensors and monitors all aspects of the safety interlocks, tube conditions, the water
cooling circuit, and so on.
The most likely problem is an interruption of cooling water from the Haskris chiller system which is not an emergency condition.
Most problems that are detected by the Bruker will result in:
- A red indicator on either the Generator Button Display or the Enclosure Display (both on the left side front panel).
- An error condition in Diffrac on the Tools tab.
- These error conditions usually turn off the x-ray tube (high voltage and tube current).
These sensors are used to shut down any system that has an error condition, so the system has built-in protection. For these reasons you should not have to take action under normal circumstances where the instrument has some problem regarding equipment parameters.
Events requiring emergency stop
The following events require immediate action:
- Electrical fire (burning smell, smoke, flame), arcing, etc.
- The Bruker goniometer attempting to drive continuously against a hard limit or any other destructive and unanticipated behavior.
If there is a real emergency, do the following:
- Press the red emergency stop button located on either side of the d8 instrument.
- Contact Paul Carpenter (Cell 314-602-9697, paulc@levee.wustl.edu)/ Jeff Catalano / Brad Jolliff immediately.
- You will need communicate what happened verbally and also write an email summarizing the specific problem and what may have caused it
to occur.
Water leaking from the Haskris building supply
If there is water leaking from the braided vinyl tubing connected to the Bruker from the building water supply (this is from the Haskris chiller in the 4th floor penthouse) then you should do the following:
- Get help immediately, if nobody is available, then continue with these instructions.
- In Commander: Stop the current XRD run using the Stop button.
- In Commander: Set the high voltage to 20 kV and the current to 5 mA.
- Wait 5 minutes if possible for the x-ray tube and electronics to finish cooling.
- Follow the instructions for Bruker Instrument Full Shutdown.
The Following Sections Are Bruker Procedures For Authorized Users Only
Bruker d8 Reboot Procedure
Bruker Reboot Procedure
STOP. These instructions are only for rebooting the Bruker d8 hardware after a change has been sent to the diffractometer, it is not part of normal operating procedures.
Some Bruker settings are saved to the instrument and a reboot is required for those changes that affect hardware components. These instructions cover the reboot procedure.
These steps are for users who have been authorized to reboot the d8.

Bruker d8 Reboot Procedure:
- Make sure the Bruker is done running a sample and that the tube has been set to standby conditions of 20kV and 5 mA.
Wait for the tube to cool for 5 minutes before rebooting because the water flow will be interrupted during this procedure.
- Press the upper Generator button to turn off the high voltage.
- Before proceeding, set the water flow valve on the wall to a higher bypass configuration so that the Haskris chiller will not go into alarm mode. This can be accomplished by setting the water flow to about 75% of the level marked on the flow gauge.
- The reboot will require the following:
The Measurement Server will need to re-connect in order to restart communications with the Bruker.
The Bruker Framework software will need to re-connect to the instrument in order to control it.
- You will not need to quit from any software on the Bruker Bragg PC (both Framework and Measurement Server will still be running but both will need to reconnect after the reboot).
- On the Bruker software application (typically Framework or Bruker.Diffrac), select the item which will save to the instrument. For example, this could be Configuration plug-in, Network menu -- Instrument Setup, with a change to an item in the window.
- Click Save to instrument to write to the Bruker hardware. This will raise a dialog window which indicates that a reboot will be necessary.
- The Bruker will reboot and the upper Generator button will show the white [|] icon. Wait about 2 minutes for the instrument to fully reboot (even though the LynxeyeXE electronics do not require this time it apparently requires about 2 minutes for the generator to be ready).
- Set the water flow back to the full level with a slight amount of bypass.
- Press the upper Generator button to turn on the high voltage supply and power to the x-ray tube.
- If the water flow is insufficient you will get a red [~] icon which will go away when the flow is higher than about 3.8 gpm.
- The lower Enclosure Display button will be white during system boot and when the high voltage is turned off.
- Open the Bruker Measurement Server program and click on the Reconnect button to restart communication with the d8 hardware. You should get a green check mark next to the instrument entry and a green text field.
- When the Measurement Server connects to the Bruker, the lower Enclosure Display button will change from white to green. This indicates normal operation.
- In the Diffrac Framework program, select File -- Connect to reconnect to the Bruker. This will take about 30sec to 1 minute.
- Inspect the Tools, X-ray Direct Water Cooling for water flow errors. The flow rate should be above 4.5 gpm.
- Go to Commander mode and initialize drives. Confirm that the drives are initialized and at correction positions, and can be driven.
- If the tube is conditioning, wait for it to finish and observe any events such as arcing that occur during the conditioning period.
- When the system is finished starting up, the Generator display button should show light radiation symbol on a dark background.
- Contact Paul Carpenter (Cell 314-602-9697, paulc@levee.wustl.edu)/ Jeff Catalano / Brad Jolliff if necessary.
Bruker d8 Cold Startup Procedures
Bruker Cold Startup Sequence
STOP. These instructions are only for turning the Bruker back on after a system shutdown.
If the Bruker has been complete turned off, the startup sequence is used to bring the unit back up to operating condition.
These steps are for users who have been authorized to turn the system on or off.
Bruker d8 Cold Startup Sequence:
- Make sure the Haskris water supply is turned on, and the valve is set to a small amount of bypass.
- On the left side panel, turn the power on by rotating the mains disconnect switch from left horizontal O (off) clockwise to vertical | (on). This turns the mains power on.
- Press the Power On | button to the left of the mains disconnect switch. This starts the Bruker internal electronics, boots the hardware computer, and opens the water flow valve that was closed when the power was turned off.
- Confirm that a good water flow is observed on the wall flowmeter and that the Haskris buzzer is not sounding in room 154.
- Wait approximately 2 minutes for the Lynxeye electronics to finish booting. (This step may be obsolete with Lynxeye XE electronics).
- On the front of the Bruker, wait for the white [|] display to appear on the upper Generator button (left side of d8), this indicates the d8 has finished booting.
- Press the upper Generator button to turn on the high voltage supply and power to the x-ray tube.
- If the system has been off for more than 2 days, it will probably go into tube conditioning mode which takes about 1.5 hours. This is indicated by the blue COND display. Tube conditioning will also be indicated in the Bruker Commander pane of the Framework software.
- The lower Enclosure Display button will be white during system boot and when the high voltage is turned off.
- Launch the Bruker Measurement Server program and confirm communication with the d8 hardware. When you open the Measurement Server window a good connection will show a green check mark next to the instrument name, some number of sent and received bytes in the I/O display, and a reconnect button. Reconnecting can be done to ensure that the computer is communicating with the Bruker.
- When the Measurement Server connects to the Bruker, the lower Enclosure Display button will change from white to green. This indicates normal operation.
- Launch the Diffrac Framework program and inspect the Tools for error conditions. Respond and correct as necessary.
- Go to Commander mode and initialize drives. Confirm that the drives are initialized and at correction positions, and can be driven.
- If the tube is conditioning, wait for it to finish and observe any events such as arcing that occur during the conditioning period.
- When the system is finished starting up, the Generator display button should show light radiation symbol on a dark background.
- Contact Paul Carpenter (Cell 314-602-9697, paulc@levee.wustl.edu)/ Jeff Catalano / Brad Jolliff if necessary.
Bruker d8 Full Shutdown Procedures
Bruker System Full Shutdown Sequence
STOP. These instructions are only for shutting the Bruker completely off. We normally leave it turned on with the x-ray tube set to 20 kV and 5 mA. Do not use these instructions unless you have specifically been told to do so.
Bruker d8 Full Shutdown Sequence:
- Make sure all measurements are complete.
- Use the Commander to set the x-ray tube to the standby conditions of 20 kV and 5 mA.
- If possible, wait at least 5 minutes for the tube to completely cool because you are about to turn off the water cooling supply to the electronics and tube circuit.
- Use the Commander to drive axes to known park positions (20 degrees, for example).
- Press the Generator button to turn off the x-ray high voltage generator (left side upper button). It will change from a light radiation symbol on a dark background to (presumably a white [|} indicating generator ready).
- Press the Standby button (left side, next to mains disconnect blade switch, lower button).
- This sets the Bruker to a condition of generator off, but the water flow circuit is enabled. The instrument
should be left in this mode if possible, because the internal water valve is shut off when the power is completely turned
off and this will cause the Haskris unit in the penthouse to go into alarm mode (very loud buzzer in room 154).
- If the Bruker needs to be completely turned off, set the water supply to bypass.
- Turn the power off (counter-clockwise to horizontal) using the mains disconnect switch on the laft side of the d8 unit.
- Contact Paul Carpenter (Cell 314-602-9697, paulc@levee.wustl.edu)/ Jeff Catalano / Brad Jolliff if necessary.
Bruker Measurement Server
Bruker Measurement Server Application
The d8 Advance has an ethernet board and communicates with the Bragg control computer using ethernet communications. This communication link is established by the Measurement Server Application. Once connected, all further control is handled by the Diffrac.Suite program package.
The Measurement Server application is started first and attempts to connect via ethernet to the Bruker d8. Once this connection
is made, all subsequent actions are handled by the Measurement Suite software.

Here the Measurement Server window shows the configuration when the server has connected to the d8 Advance, and is currently idle with no active jobs.
- The Bruker d8 (named Washington University in St Louis) has the check box selected in the Control column and once connected can be controlled by the Diffrac.Suite program.
- The green check mark in the Status column indicates that communications have been established.
- The Job Schedule Status field is colored green and contains the keyword Idle when the instrument is not actively running a job.
- The Network Packets (selected instrument) shows packet traffic between the PC and the Bruker d8.
- It is normal for the Reconnect button to be colored red.
Confirm Measurement Server is running and PC is connected to the Bruker d8 Advance
On the Bragg control computer confirm that the Measurement Server is running and connected to the Bruker d8:
- First determine if the Measurement Server is running: If there is an icon for the Measurement Server on the PC taskbar, then it has been launched but may not be connected to the Bruker d8. If the icon is there but the Enclosure Button on the d8 is not colored green, then the server is not currently connected.
- If there is no Measurement Server icon on the task bar, launch it from the Start Menu on the PC, and wait for it to connect. This takes about 1 minute and when it connects the Enclosure Button on the d8 will then turn green.
- On the lower right task bar, right-click on the Measurement Server icon and select the Status window item.


- When the Measurement Server window opens, confirm that there is a green check mark under the Status column for communications with the Bruker, and that the Job Scheduler Status text field is green with the word Idle. This all means
that the computer is communicating with the Bruker and no active measurements are in progress.
- If the Status field is not green and/or communication has been somehow interrupted, you can click on the Reconnect button to restart the ethernet communications. If this does not work get help.
- Close the Measurement Server window.
XRD Lab Computers, Software, Databases, and Scheduling
The following information covers the available instrumentation in the lab, and the computers that are used for data acquisition and data processing.
Scheduling Resources in the XRD Lab
There are currently two XRD systems in the lab, the Rigaku and Bruker XRD instruments, and additionally the new XRD workstation. The scheduling of these resources is done as follows.
- Scheduling of the Bruker XRD includes exclusive use of the Bragg PC. You can do everything except search match using the ICDD PDF5+ database, which is on the Rietveld computer only.
- The Rietveld XRD workstation is available for use at any time (this includes nights and weekends since there is no safety issue related to ionizing radiation), but priority will be given to users of the instruments that require data processing during their XRD session (such as search match using the ICDD PDF5+ database).
- Scheduling of the Rigaku XRD includes only use of the Rigaku PC for Datascan data collection. Jade is no longer installed on the Rigaku PC so you must also schedule time on the Rietveld XRD workstation if you need to do Jade processing during your Rigaku session. So you need to reserve both the Rigaku and the XRD workstation at the same time using the scheduler.
Bragg PC -- Bruker d8 Main Control Computer
The Bragg PC computer is used for both data acquisition using the Bruker d8, and for processing of data. The main capabilities are listed below.
- Bruker Measurment Server. This is the server program that connects to the Bruker d8 diffractometer to perform all data acquisition.
- Bruker.Measurement Program. This is the Framework software that is used to run Wizard, Start Jobs, Commander, etc. and handles all the actual running of samples.
- Storage of User XRD Data Files. The Bragg computer is used for the storage of both Experiment and Results files. The Results files are processed using Diffrac.Eva, Topas, and any other programs. The Results .raw data files can be processed on Bragg or from Rietveld using the network drive to Bragg.
- Bruker.Eva. This is the program used to view and process XRD scan data. It is used to perform search-match using whatever database is installed on a given computer.
- Database Files on Bragg. Only the Crystallography Open Database (COD) database is installed on Bragg. The COD database is searchable using Diffrac.Eva and is in a format that Eva can use, either by pattern searching or by lookup from the name of the phase.
If you need to use the ICDD PDF5+ database, you will need to reserve and use the Rietveld computer.
- Topas Rietveld Refinement. The Bruker GUI version of Topas is installed on Bragg, in the C:\Topas directory. There are subdirectories that contain structure files (STR that can be loaded into Topas for refinement), and some CIF files that can be loaded (generally with some user input to correct cation names etc.) for refinement use.
- Diamond Crystal Structure Software. We have a site license for Diamond in Earth and Planetary Sciences. This software is very nice for viewing the crystal structure and XRD pattern. There is a tutorial in the Diamond directory, and it can import CIF structure files.
- Utility programs. There are a number of utility programs including Bruker File Exchange, PowDLL, and others.
Rietveld PC -- XRD Workstation Computer
The Rietveld PC computer is a high performance PC with 16 gb RAM and high speed SATA6 hard drives. The main capabilities are listed below.
- Access to User XRD Data Files. The Rietveld computer uses network drive B to access the Bragg User Data folder which contains your XRD Results files. You should process your .raw files in place (i.e., remotely from Rietveld) on the Bragg computer using Bruker.Eva. This is the program used to view and process XRD scan data, and to perform search-match using whatever database is installed on a given computer.
- Database Files on Rietveld. The Rietveld XRD workstation has both the ICDD PDF5+ and COD databases installed.
You can perform search match procedures from Diffrac.Eva using either the PDF5+ or COD databases and a user database, but not both at the same time. You can also do search match using line based or whole pattern methods from the Jade program.
- Topas Rietveld Refinement. The Bruker GUI version of Topas is also installed on Rietveld, in the C:\Topas directory. There are subdirectories that contain structure files (STR that can be loaded into Topas for refinement), and some CIF files that can be loaded (generally with some user input to correct cation names etc.) for refinement use.
- Diamond Crystal Structure Software. We have a site license for Diamond in Earth and Planetary Sciences. This software is very nice for viewing the crystal structure and XRD pattern. There is a tutorial in the Diamond directory, and it can import CIF structure files.
- MDI Jade. The Jade program is very good and complementary to the Bruker software. The whole pattern search match is very good and allows searching on the full pattern followed by the difference or residual pattern. Jade also has crystal refinement, structure viewing, and XRD pattern simulation capabilities. It does not have Rietveld or quantitative analysis capabilities and you should use Topas for that.
- GSAS and EXPGUI. These are installed on Rietveld and are available for you to use.
- Fullprof. Installed on Rietveld and available for you to use.
- Utility programs. There are a number of utility programs including Bruker File Exchange, PowDLL, and others.
XRD Resources on Rietveld PC -- XRD Workstation Computer
The Rietveld PC computer has a directory of resources for XRD users, located in the XRD Users directory on drive D. The materials are listed below and are available for you to copy; please do not move them from their folders.
These resources include journal papers, powerpoint presentations, tutorials, and other materials that are topically centered on Rietveld refinement using the Topas program. There is also information about XRD quantitative analysis and crystal structure determination. Be aware that these materials are being organized and may be reclassified as time goes on.
- Location of XRD User resources. On drive D the XRD Users folder contains the directory "XRD Resources". This directory is accessible only from the computer console.
- Bruker Good XRD Practice. Presentations covering crystallography, general XRD procedures and technology, and applications.
- Crystal Structure Determination. Performing crystal structure determination using x-ray powder diffraction data.
- ICDD Tutorials and other documents. Copies of tutorials and information from the ICDD web site, covering use of the PDF5+ utility program, searching the PDF5+ database including data mining techniques, and applications to mineralogy and the pharmaceutical industry (for example). The ICDD web site also provides access to papers from the annual Denver X-ray Conference which focuses on XRD (including Rietveld refinement)and XRF analysis.
- Powder X-ray Diffraction. General papers covering powder XRD and advances in the field.
- Quantitative Analysis by powder XRD. QA is the measurement of the weight percent quantity of phases in a multiphase mixture (e.g., a soil, rock powder, chemical reaction series, etc.). This procedure is performed using Topas in our lab. There are papers covering the details of QA and the reports on several round robin studies where phase mixtures of varying analytical challenge were analyzed by laboratories and a summary of the results.
- Rietveld refinement. General papers on Rietveld refinement and advances in this technique.
- Meetings and Workshops on X-ray Powder Diffraction. Several meetings and workshops on powder XRD have resulted in presentations and booklets which are valuable sources of information on crystallography, Rietveld refinement and applications, and structure determination.
- Durham School - John Evans. The materials from the Durham school include information on aspects of crystallography and powder XRD but also have tutorials which have been set up for the academic version of Topas. See the web link for more information. Most of the tutorial examples are in the Topas4-2 directory on Rietveld (see the Tutorials and other directories).
- XRD Structure Databases. Papers with information on the CIF format, the Crystallography Open Database (COD), and the Mincryst files that are in a format readable by the Jade program.
- XRD Textbooks and materials. The electronic documents, problems, and images from Fundamentals of Powder Diffraction by Vitalij K. Pecharsky and Peter Y. Zavalij are available. The library has a copy of this textbook.
Sample Preparation for an XRD run
- Obtain representative sample.
- Grind sample to ~10 micron grain size, best grind is in acetone or isopropanol (i.e., not dry).
- Approximately 80-100 mg of powder required.
- See the next section for information about Bruker sample holders. We use machined aluminum holders for normal sample powders, backloading holders for samples with preferred orientation issues, and the MTI zero background Silicon holder for small sample quantities.
- Load sample into the appropriate holder--please try to not scratch the holder. Sample should completely fill the round cavity and be exactly flush with the top surface.
- Sample must be flush with top surface of the holder:
If sample is above surface of holder then positive z-axis
displacement error will move the XRD spectrum to higher angles
If sample is below surface of holder then negative
z-axis displacement error will move XRD spectrum to lower angles.
- Avoiding preferred orientation by excessive agitation of sample.
- Hygroscopic or other reactive powders may be sealed using Saran wrap, Kapton x-ray film, etc. to reduce reaction with the atmosphere.
- Other capabilities include RIR method (use 1 micron Al2O3 as internal reference standard, acquire standard XRD spectra, use FullPat or similar program to fit sample spectra to standards).
- Place the round sample holder into the Bruker sample assembly--do this outside of the Bruker instrument hutch and on the counter
so that if you spill sample it can be cleaned up. Clean up any residual powder from the holder before you put it in the Bruker sample holder.
Sample Holders for the Bruker d8
The Bruker d8 Advance is configured with the X-ray tube in line mode. This means the X-ray beam is 1" in y-axis and approximately 2 mm in x-axis direction, so that when the sample holder is rotated, a circular 1" area is irradiated by the X-ray beam.
We have several sample holder types for the Bruker d8, including powder sample holders provided by the lab, and thin-film sample holders which your research group will need to have made (Chemistry machine shop, Jim Linders, jlinders@wustl.edu). These are described as follows.
Bruker Sample Holders for Powder Samples

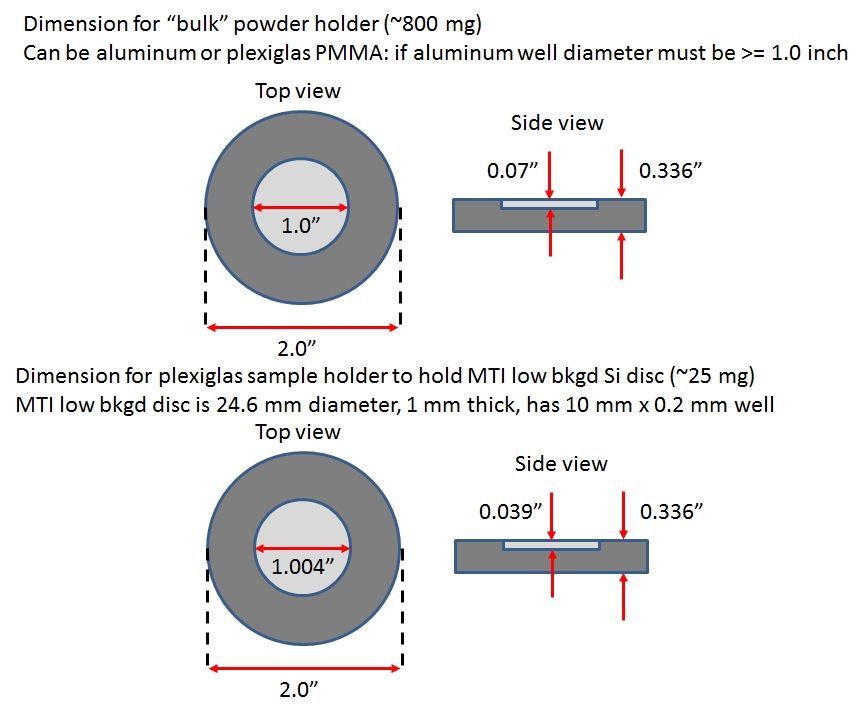
- Bulk powder sample holder (machined aluminum).
This aluminum holder has a 1-inch diameter well which holds approx. 800 mg of sample. It should be used for most sample powders if you have that much material.
Powder should be put into the well and gently smoothed to a continuous surface with the aluminum holder by using a glass slide.
- Back loading plexiglas sample holder (plexiglas). This holder is used for samples that exhibit preferred orientation problems.
Procedure for using the back loading holder:
- The holder should be placed face-down on a weighing sheet on a hard surface, then place the plexiglas funnel into the sample holder.
- Add sample powder and use the plexiglas pestle to pack the powder so that it will not fragment. Remove the plexiglas funnel.
- Place the black metal insert into the holder to back the sample powder.
- While holding the weighing paper, flip the holder upright. The sample is now ready to run on the instrument.
- Zero background Silicon sample holder (1-inch/25.4 mm diameter with 10 mm x 0.2 mm central cavity).
For small sample quantities you will need to use a Silicon low background sample holder that is sold by MTI, and which holds approx. 25 mg of powder. We have several plexiglas holders that have been machined specifically for the MTI Silicon holder. Place the MTI Silicon holder in the plexiglas holder and add sample powder, then load the sample holder into the Bruker assembly.
To purchase the MTI holder see MTI link here. Note, do not purchase the 1-inch holder that has no central cavity because our plexiglas holder is designed for the holder with a recessed cavity.
Bruker Sample Holders for Thin-film Samples
Please note that your research group will need to have these holders machined.
- Because each thin film sample has different XY dimensions and Z thicknesses, sample holders will need to be custom fabricated. Both aluminum and plexiglass can be used for the holder material.
- The X-ray beam is 1" in length by ~ 2mm in width. If your thin film package has an XY dimension less than 1", you will need to use a plexiglas holder or mask off the area of an aluminum holder to avoid diffraction peaks from the aluminum exposed to the X-ray beam.
- The thin film package must also be mounted in the holder so that the top surface is flush with the holder. If it is above the surface a positive Z-axis displacement error will be observed (diffraction peaks at higher angles than expected), and if below the surface the opposite will be observed.
- The photo below shows a top view of several holders machined from aluminum, with the well depth indicated for typical thin-film packages to be measured on the diffractometer. The 0.625 mm well is appropriate for use of 5-inch Silicon wafer stock which is 0.625 mm thick. Remember that you need a deeper well for the substrate plus whatever thickness the thin-film assembly adds for the total Z-axis thickness. Use aluminum foil or other shim material to raise the package to the required Z-axis position.
Thin-film sample holders (machined aluminum).

Policy Statement for Use of Bruker d8 Advance
The Earth and Planetary Science Bruker d8 Advance is made available to selected members of the EPS department, Washington University, other non-profit organizations, and commercial clients. Users are permitted to use the instrument subject to the following requirements:
- Mandatory user training conducted by EPS staff. This consists of two parts:
Part 1: 3 hour demonstration and discussion of Bruker d8 operation, Framework data collection, EVA and PDF5+ software, and Topas Rietveld refinement.
Part 2: Demonstration of ability to follow Bruker d8 web procedures, perform sample exchange, and run samples. Training is not complete until part 2 has been demonstrated in a separate session.
- You must maintain a signed entry in the lab blue book with a current EHS lab safety training date.
Use of the XRD lab without a current EHS certificate and blue book entry is unauthorized.
- See below for how to access the WUSTL EHS and Learn@Work web sites.
- The procedures on this web page are your annual lab-specific user training and you are therefore required as part of your annual EHS training to review these procedures.
- Users who do not follow procedures in the on-line manual, cause problems regarding necessary care during use of the instrument, software procedures, and lab cleanliness will have their check-out revoked.
Bruker d8 XRD Training
Please note that users are trained approximately once a month in group sessions only. Be prepared to spend approximately 3 hours in these training sessions and come prepared to use the software by reviewing the procedures in this web document including the Bruker pdf manuals. You will be asked to demonstrate your knowledge of these procedures and your ability to follow them during and after the training session. If you do not demonstrate adequate knowledge you will not be able to use the instrument.
Each research group has a "main user" that is the person responsible for helping new users by running samples during the period between training sessions. Contact Paul Carpenter to be added to the list for the next training session. You cannot be trained by another user.
In order to add you as a new user, Paul Carpenter needs the following information:
- Your first and last name, and email address.
- Your advisors first and last name, and email address.
- Which department at WUSTL or other affiliation, and the first 4 digits of your department charge code (not your campus box number).
WUSTL Environmental Health and Safety (EHS) Training and Verification From Compliance Web Page
A current EHS training certification is required to use any laboratory at WUSTL.
Use of the EPS XRD lab is not authorized without a current certification and entry in the lab blue book.
If you have not taken the laboratory safety EHS training, go to the EHS web site and see the instructions for EHS training using the Learn@Work web site:
Environmental Health and Safety web site
Learn@Work web site
To obtain proof of your EHS on-line training, you can access your electronic information on the Learn@Work web site:
- Log in to the Learn@Work web site using your WUSTL key.
- If you have completed the EHS Lab Safety Training, this will be shown in the list.
- Click on the link for EHS Safety Training Certification.
- You only need the date of EHS training to enter in the EPS Room 152 blue book. We do not need your certificate.
- If the EHS date is more than one year old, you are not in compliance and you need to take your annual EHS training.
Laboratory Operating Hours
The XRD lab is available for use during normal weekdays from approximately 9am-5pm. It is also necessary for Paul Carpenter or a designated person to be available for supervision of your time in the lab. The lab is closed on weekends and holidays.
The building and hall require electronic key card access so that lab is not available on nights and weekends except to EPS personnel. Please remember this when scheduling time on the calendar. The XRD laboratory is kept locked because it is an environment where ionizing radiation is used. For routine users a key can be obtained for entry into the lab. Please use one key for your research group (let Carpenter know if you are a user in a new group that does not have a key).
Current fee structure established 2024 after no increase in rates since 2013.
- The EEPS XRD Facility is a WUSTL cost center and is required to cover all lab costs from user fees. The university does not provide direct support.
- Maintenance costs include Bruker on-site repairs, X-ray tube and parts replacement, ICDD PDF5+ database subscription, and other costs.
- All fees are on a per hour usage basis.
There is a 0.5 hour minimum instrument usage charge per session.
- There is a nominal one hour instrument and one hour operator charge for XRD training.
- Users that do not complete their final checkout after one year will need to go through XRD training again.
- Instrument time is determined from run time recorded in the Bruker user database.
- Use of the Rietveld XRD workstation, software, and ICDD database access are currently free.
Operator assistance and training will be charged based on required assistance by individuals.
- Operator time is charged thereafter as determined by Carpenter to reflect both per-user assistance and may include a portion of total effort.
- Material fees are chaged for:
- Use of laboratory Si zero background MTI holder, charged on a per session basis.
Replacement cost is assessed if the holder is broken or removed from the lab.
|
Instrument Rate
|
Additional Operator
as necessary
|
Material use fee
|
Earth, Environmental, and Planetary Sciences
|
$36
|
$36
|
$20
|
| Washington University (non-EPS) |
$36
|
$36
|
$20
|
Other Universities and Non-profit Organizations (external)
|
$48
|
$48
|
$25
|
Commercial Users
|
$102
|
$102
|
$30
|
New! WUSTL Box: XRD Resources Folder Powder X-ray Diffraction
The link below should allow you to access the XRD Resources folder on the WUSTL Box site. There is a large collection of powerpoint files, journal and other pdf documents, and video presentations on many aspects of X-ray powder diffraction analysis. There are also several XRD short course documents that can be used to improve your knowledge of XRD analysis. Enjoy!
Link to Box folder: WUSTL Box Folder: XRD Resources
Powder X-ray Diffraction
This section summarizes the technique of Powder X-ray Diffraction and provides some links to further information, see also the reference list for more complete treatment.
Important points:
- Crystalline solids are structurally composed of periodic arrays of atoms that have differential X-ray scattering probability.
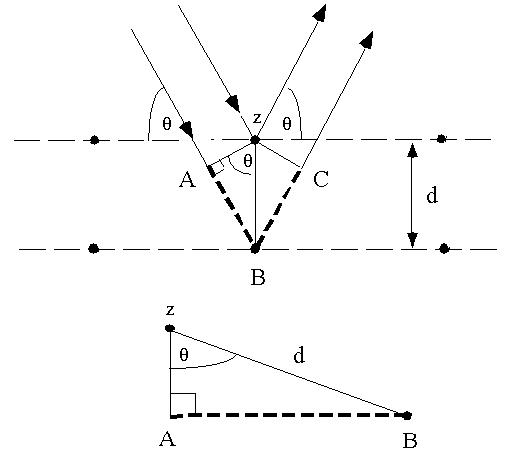
- When placed in an X-ray diffractometer and exposed to monochromatic X-rays, the X-rays are diffracted according to Bragg's law:
n * λ = 2 * d * sin(θ)
n is the order of diffraction
λ is the wavelength of X-rays (on our instrument it is Cu Kα, units are in Angstroms)
d is the interplanar spacing between layers in the crystalline structure, units also in Angstroms
θ is the angle of the diffracting X-rays to the layers of atoms which cause the constructive interference of the X-rays

- The interpretation of Bragg's law is that whenever the path length of diffracted X-rays is equal to twice the d-spacing of the layer of atoms (multiplied by the sine of the diffraction angle), then constructive wave interference occurs and a diffraction peak is observed. In the figure below, the distance AB is d*sin(θ) and for constructive interference the X-rays have a path length of AB + BC which is 2*d*sin(θ).
- In powder X-ray diffraction, a randomly oriented, finely ground powder (~ 10 microns) is required for analysis. The powder can be in the simplest case a single crystalline phase (e.g., quartz), or a mixture of phases (e.g., quartz, calcite, dolomite). As the number of phases or diffraction peaks increases it becomes more difficult to identify all phases.
- Below is an X-ray powder diffraction plot collected on halite (NaCl) using a Cu Kα X-ray tube source. The data are collected by progressively increasing the angle between the sample and both the X-ray source and detector (which are at the same angle to the sample for a θ-θ instrument) and counting the detector at each angle. For this sample the diffractometer was driven from about 17.5 to 60 degrees 2θ. The peaks on the diffraction plot show the angles where Bragg's law is satisfied, i.e., the angles where n*λ = 2*d*sin(θ) for the NaCl structure. The regions of background scattering in between the peaks are angles for which n*λ does not equal 2*d*sin(θ). Note that the diffraction peaks are related to fundamental atomic planes in the NaCl lattice and that the plane with largest d-spacing is diffracted at the lowest angle while those planes with smaller d-spacing are diffracted at higher angles. Also, the d-spacing for the (222) reflection is half of that for the (111) reflection thus indicating the relationship between Miller indices of a plane and the d-spacing for that plane.
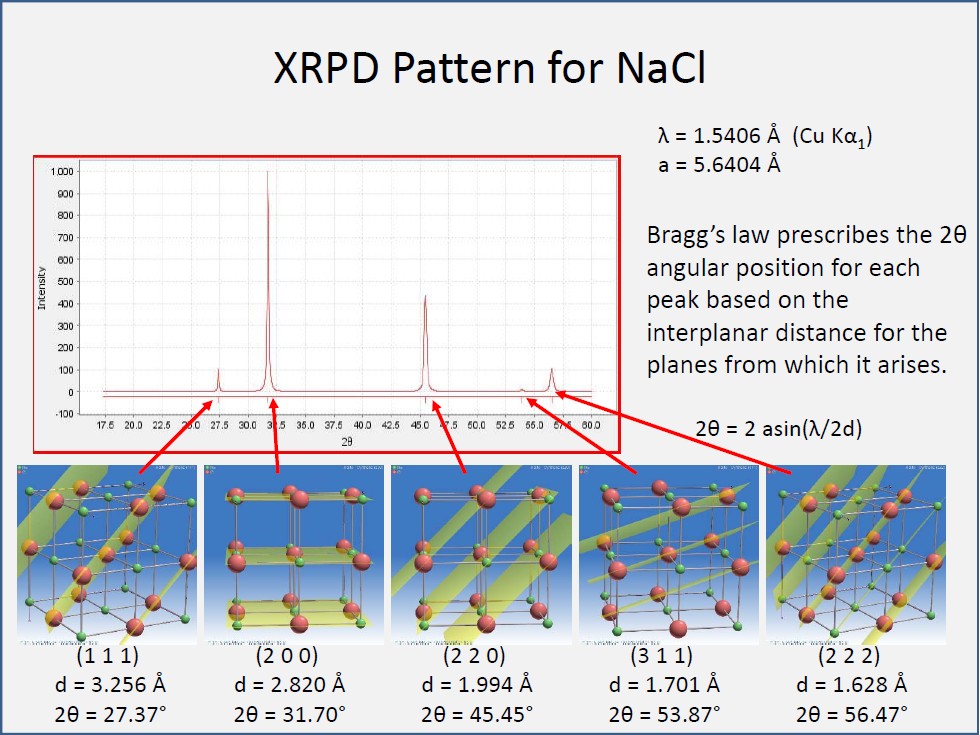
- For any powder diffraction sample, the data are collected in this same way, and the peaks are identified by a software peakfinding routine. Because each 2θ position is equal to a specific d-spacing, the peaks are equivalently represented by either 2θ position or d-spacing in Angstroms. For the NaCl pattern, the (200) plane has the highest diffraction intensity and represents the atomic layer in the NaCl structure with highest atom density, while the (311) plane has the lowest intensity and represents a layer with lower atom density.
- The actual X-ray powder pattern for NaCl is reduced to a "D-I" list which is the list of each diffraction peak in terms of the d-spacing and relative intensity compared to the most intense peak observed on the pattern. For NaCl the (200) is most intense and is labelled the 100% peak, and all other peaks are scaled relative to the 100% peak.
- The Search-Match (SM) procedure is the method used to identify an unknown material. The SM procedure takes the D-I list from the sample and compares it to all ICDD "cards" in the electronic database sub-file that we have chosen. For NaCl we presume that it is inorganic and likely a mineral so we would search those smaller files. The search in principle can include several hundred thousand cards. There are several methods of SM; the line-based procedure takes the intensity-ranked D-I list and compares it to that of each card. A whole-pattern SM method matches the pattern of the sample to that obtained by generating a diffraction pattern from the known D-I list of cards in the database.
- The ICDD card 04-016-2944 for NaCl (one of many cards for NaCl) is shown below. The card contains the D-I list and an image of the experimental diffraction pattern for comparison. It is a simple and easy match due to the simplicity of the NaCl structure and the absence of any other phases in the sample. Notice that the ICDD card has the Miller indices of the peaks identified and also has cell data and atomic position coordinates that are needed for Rietveld refinement.

- We are able to easily and quickly identify the "unknown" material as NaCl because a pattern (actually many experimental patterns) exists in the ICDD PDF5+ database.
- If the unknown material was in fact a new structure, the search-match procedure would not find a match because no material with the same peaks and intensities (the D-I list) would be an exact match. We would need to first determine the unit cell parameters of the unknown material, then determine the HKL indices of the diffraction peaks (this is called indexing). This procedure also tries to match space groups based on allowed vs. disallowed peaks in comparison between the observed material and candidate space groups. For a cubic material like NaCl it is easy, for a triclinic phase it is much more difficult. The space group identifies the symmetry elements of the crystal structure but does not explicitly determine the positions of the atoms in the unit cell. Crystal structure analysis is a set of procedures that attempts to place the atoms in positions in a way that is compatible with the observed diffraction pattern. The result is a complete description of the crystal structure.
Summary of Powder X-ray Diffraction Technique
The technique is summarized as follows:
- Sample preparation and data collection
A powdered sample is prepared and data collected by scanning 2θ while exposing the sample to monochromatic X-rays. The diffraction data obtained represent the crystalline structure of either single or multiple phases in the powder.
- Generate D-I list
The peaks in the diffraction pattern are converted to d-spacing and relative intensity compared to the most intense peak observed. In general, phases with high symmetry such as cubic structures have relatively few peaks, and phases with low symmetry such as monoclinic and triclinic phases have many diffraction peaks. These inherent characteristics affect the success of phase identification especially minor phases present with a major low-symmetry phase.
- Search-Match Procedure
The D-I peak list is used by the Search-Match procedure to search the ICDD database for phases which have ideally the same d-spacing and relative intensity values. For data collected over a wide enough 2θ range so that a sufficient number of peaks are observed, and using a purified phase that exhibit no minor or trace phases, one can expect a short list of candidate matches to compare with the sample diffraction spectrum.
Scans with only a few peaks result in more possible matches which are almost all incorrect; scans with many peaks yield perhaps one or two matches. The success depends on the data range collected, degree of crystallinity of the sample, purity and single-phase nature of the sample, and other specifics related to crystallography.
The D-I list or equivalent pattern is thus a fingerprint of the sample and can be used to identify unknown crystalline materials that can be organic, inorganic, mineral, metal, ceramic, etc.
The identification of phases in a sample is made using Search-Match procedures. This step is necessary prior to Rietveld refinement.
- Rietveld refinement Summary
Rietveld refinement is a method where an XRD pattern is calculated in comparison to the experimental pattern, and parameters are adjusted in order to minimize the residual difference between the calculated and experimental patterns. These parameters include cell dimensions and angles, peak intensities, crystallite size, and phase abundance for multiphase samples. It is a whole-pattern calculation in typical application where the least-squares fit of the calculated pattern is based on all channels in the experimental pattern rather than single peaks (although this can be done as well). It primarily uses peak fitting and optimization procedures and is applied to single-phase and multiphase materials. It requires that all phases have been identified by Search-Match procedures previously as this function is not part of the refinement software.
In particular:
- Refinement can mean adjustment of parameters such as fitted peak intensities and 2θ positions, cell dimensions, atomic positions, crystallite size, etc. There can be restraints (progressive resistance to change in a parameter) and constraints (outright limits to change beyond a specified range) applied to values actively being refined, or values can be fixed (such as instrumental constants). Values can be refined initially then fixed as other parameters are adjusted.
- Values are more conveniently obtained via refinement compared to the use of specialized software previously used for procedures such as indexing.
- Non-linear least squares is used for spectrum processing and a whole-pattern residual is calculated to evaluate goodness of fit.
- Instrumental parameters are used to calculate the effect of X-ray source, instrumental resolution, axial divergence, sample displacement, zero error, and other factors. These parameters are typically fixed.
- Modelling parameters are selected, such as Gaussian vs. Lorentzian peak shape analysis used for peak deconvolution. Multiple peaks are used to deconvolve complex peak shapes in the experimental pattern, and/or to describe strain effects for example.
- Sample parameters that are refined include unit cell and space group, atomic coordinates, site occupancy, thermal parameters, preferred orientation, crystal size, strain, and others.
- The Bruker Topas software uses .str structure files exported from the ICDD PDF4/5+ database as input for refinement processing. For most materials a .str structure file should be used and can be edited if necessary. It is also possible to use .cif files as input, and to create structures within Topas. The software can be run in GUI or launch mode where it uses the Topas script language directly.
- Refinement of patterns acquired on multiphase samples (i.e., rocks and soils) uses multiple .str structure files appropriate for the phase mixture and produces a weight percent analysis of the sample. This is known as quantitative analysis. Typically, the inventory of phases in a multiphase sample is determined by Search-Match, then refinement is used based on .str structure files for those phases. Inspection of the residual may reveal missed phases such that the experimental pattern has peaks not produced in the calculated pattern. This drives subsequent Search-Match and refinement iterations.
- Rietveld refinement of XRD on powder samples has effectively replaced single-crystal methods based on convenience. It also provides rapid determination of parameters for many materials. It is not as good for some aspects such as determination of temperature factors.
- For identification of true unknown materials, Topas can perform indexing and structure solution using refinement. These procedures require understanding and iteration. Routine refinement using .str files is straightforward in comparison.
- See the lab XRD Resources, Bruker Topas manuals, and web-based materials for further information.
- Quantitative Analysis
The identification of multiple-phase mixtures and the estimation of their weight percent in the mixture is called quantitative analysis. Rietveld refinement works well assuming you have the structure data for all phases. There is not a linear relationship between weight percent of a phase and peak height in the XRD spectrum; traditionally a calibration curve was used based on known mixtures. Quantitative analysis using Rietveld refinement has replaced that procedure. Note that the whole pattern residual and error calculation in Rietveld refinement refers to peak fitting statistics and the residual between the calculated and experimental pattern. The accuracy of quantitative analysis relies on measurement of known mixtures and a comparison of the calculated vs. known phase abundances. Determination of amorphous content can be estimated or measured by addition of a known amount of a crystalline material, usually NIST Al2O3 SRM.
- The XRD technique requires a sufficient amount of powder to collect data, and the use of insufficient powder results in poor sensitivity (this can be remedied by collecting data on a Si low-background sample holder). As well, collection of data on a multiphase sample in which there is a trace constituent (i.e., less than 2%) shows relatively low sensitivity unless that material is a high-Z phase which efficiently scatters X-rays. The technique is therefore not a trace analysis method like ICP-MS or other chemical techniques but rather a structural method.
Links for Powder X-ray Diffraction
Here are links to information about Powder X-ray Diffraction and related topics.
- Wikipedia articles:
- Powder diffraction
- Bragg's law
- X-ray crystallography
- Rietveld refinement
- Links to websites with good discussion of X-ray powder diffraction
- XRD method
- ICDD website tutorials and other materials. The tutorials cover powder diffraction and use of the ICDD PDF5+ database for which we have a subscription, and the PDF5+ database appliction that is very good for searching ICDD card data. You can also find other educational materials on the site.
- ICDD Web Site
There are general references to powder x-ray diffraction and more modern references which highlight the widespread use
of Rietveld refinement in processing powder patterns.
All of these references are currently listed in the Washington University Library catalog. Here they are ordered by date.
- X-ray diffraction procedures for polycrystalline and amorphous materials The comprehensive text on x-ray diffraction.
Harold P. Klug and Leroy E. Alexander. 2d ed. New York, Wiley 1974
- Modern powder diffraction MSA Short Course volume.
D.L. Bish and J.E. Post, editors. Washington, D.C. : Mineralogical Society of America, c1989.
- X-ray diffraction A Dover book with theoretical aspects of x-ray diffraction.
B. E. Warren. New York : Dover Publications, 1990.
- Introduction to X-ray powder diffractometry
Ron Jenkins and Robert L. Snyder. New York : Wiley, 1996.
- Applications of synchrotron radiation to materials analysis
edited by H. Saisho and Y. Gohshi. Amsterdam ; New York : Elsevier, 1996.
- X-ray diffraction and the identification and analysis of clay minerals
Duane M. Moore, Robert C. Reynolds, Jr. 2nd ed. Oxford ; New York : Oxford University Press, c1997.
- A practical guide for the preparation of specimens for x-ray fluorescence and x-ray diffraction analysis
Edited by Victor E. Buhrke, Ron Jenkins, Deane K. Smith. New York : Wiley-VCH, c1998.
- Powder diffraction : theory and practice
(Note that this appears to be available as a Google book)
Edited by Robert E. Dinnebier, Simon J.L. Billinge. Cambridge, UK : Royal Society of Chemistry, c2008.
- Fundamentals of powder diffraction and structural characterization of materials An excellent modern text with electronic materials
Vitalij K. Pecharsky, Peter Y. Zavalij. 2nd ed. New York : Springer, c2009.
The Bruker d8 X-ray Diffractometer is supported by NSF grant funds and user fees. Please include the following information in the acknowledgements section
of published papers that use data acquired on this system. You can use the following statement:
Use of the Bruker d8 Advance X-ray diffractometer in Earth and Planetary Sciences at Washington University in St. Louis is supported by the National Science Foundation, award no. NSF EAR-1161543.
Laboratory Safety Document -- Appendix 4 of Blue Book
X-ray Diffractometer Lab (EPS Rooms 152-153)
Use: In this lab, we work with an instrument that generates X-rays. Exposure to X-rays is harmful
to you even though you can not see them or necessarily feel their effects. Our instrument has an annual radiation
survey and the results are in the laboratory blue book for the current and past year surveys. The sheilding
and safety interlock system permit us to work without the requirement of dosimetry badges. During normal operation,
you will not be exposed to harmful levels of radiation.
It is important for you to follow the standard operating
procedure and placards on the instrument during sample exchange. Failure to do so will result in you being
banned from the laboratory.
As with any radiation source, you should limit exposure time and maximize shielding and distance from the
X-ray source. Note that the exposure limits during pregnancy and for minors are much lower than for routine workers. It is
therefore not recommended to use any x-ray source during pregnancy or to bring children into the lab. For information
concerning typical exposure levels see http://www.new.ans.org/pi/resources/dosechart/
The main concerns for safety in this lab lie with sample preparation and with cleaning (see below).
For sample prep, we work mainly with powders. Most of the powders used in this lab are harmless.
If your samples are harmful, you need to take appropriate precautions in handling them to prevent
contamination of the lab, bench tops, and instrument. Acetone and Ethanol are present in the lab
for after-analysis clean-up. The instructions here are meant to supplement the University’s Chemical
Hygiene and Safety Training. After this instruction, please sign the Bluebook for the Lab indicating
that you have received the annual Lab training.
OSHA requires every laboratory to have laboratory-specific training and documentation.
- Use of Volatile Organic Chemicals, Corrosives, Flammable Materials, Epoxies, and Toxins
- Use these chemicals only in the chemical fume hood.
- Wear appropriate eye protection, foot protection, lab coat, and gloves.
Long pants and closed-toe shoes are required; sandles are not permitted (this lab has the potential for exposure to broken glass and high-voltage sources are prsent).
- Store segregated by hazard class and label as Flammable, Corrosive, or Oxidizer.
- Clean up solvent spills using a towel or other absorbent.
- Working with Powders
- If your samples are harmful (e.g. if your samples contain Arsenic, Barium, Chromium, Lead, Mercury, Nickel, Osmium, Selenium or Silver), you need to take appropriate precautions in handling them to avoid contaminating the lab or bench tops.
- If you use the mortar and pestle with your samples, clean it completely using the St. Peter’s sandstone to remove any staining.
- If you spill, there is a spill kit in the prep lab, if needed.
- Dispose of waste properly (i.e. put in plastic baggy and remove as hazardous waste through Environmental Health and Safety).
- Labelling Powders
- If you leave your samples in the lab for any period of time, they must be labeled properly.
- The label must have your name, the date, and what the sample is composed of.
- If the sample is harmful, it should be in a secondary container that you will supply.
- Proper Disposal of Sharps and Broken Glass
- Discard razor blades in Sharps container.
- Discard broken glass in broken glass box.
- Use of the Chemical Fume Hood (room 153)
- The fume hood should not be used as a storage area for chemicals or equipment.
- All containers must be capped when not in use. Evaporation of chemicals is prohibited.
- Work at least 6" inside the hood with a hood sash opening of approximately 14 inches.
- Use of Carcinogens
- Carcinogens are not permitted in this lab during normal procedures and carcinogen training is not routinely provided.
- If your samples contain carcinogens, please notify lab personnel.
- Consumption of Food and Beverage
- Consumption of food and beverage is not permitted in the sample prep lab (153).
- Beverages may be consumed in the main XRD lab (room 152) as long as the beverage is not set on the XRD console or near computer equipment.
- The lab is equipped with an eye wash and 2 showers should you need them
- Flush your eyes for as long as you can stand it.
- Contact your supervisor or a health and safety officer to report the incident and to receive further instructions.
- Fire extinguishers
- The lab (152) is equipped with a fire extinguisher.
- Familiarize yourself with its location and use.
- If you think of something that has been left off this list, please report it to the laboratory manager so that it can be incorporated in the next version of this training.
Excel User Sheet
- If Excel is not launched, click on the Excel User Sheet icon to launch it.
- Enter the date, last and first names, email address, advisor last name, first four digits of charge code, and user code.
- In the Excel sheet, enter the number of hours that you have used the Bruker (this can be determined from the database time stamp but we also ask you to provide an estimate of usage).
XRD Utility Programs
The following procedures are useful for XRD information.
MDI Mineral Program
The MDI Mineral Program can be used to find information for minerals. Launch the program and either scroll down the
list or enter the mineral name. This will load information about that mineral. A pictograph of the diffraction peaks
is shown at the upper left; hover the mouse over this graph to determine the 2 theta position of peaks and the start
and end values for a routine scan. The data also lists at least one ICDD card for this mineral, which you can use in Jade
to force a comparison in the Search Match display. This program is a quick and easy utility to determine x-ray information.
PDF5+ Utility Program for ICDD Database
The PDF5+ program is the front end utility that can be used to call ICDD card data from the ICDD database. We get the DDview+
utility with our subscription to the PDF5+ ICDD database. There is a help file that you can copy that covers routine proceduures
with the DDview+ program. There are also web based tutorials on the ICDD web site that include DDview+ information, see
http://www.icdd.com for more information.
Here are some short instructions for retrieving ICDD card information:
- Launch PDF+ from the desktop shortcut and wait for it to load; it requires a current version of the ICDD subscription to work.
- Use File Open Card to retrieve a specific card from the database.
- To find cards for an element or compound, use the periodic table mode to call up ICDD cards for specific element lists.
Note that you need to specify the correct boolean test to discriminate cards which contain only the selected elements
or elements in addition to those selected.
- When the list is loaded, double click on the card number to display the card data.
- You can use the MWSnap screen capture utility to capture any image in the PDF+ display.
- Additional constraints can be applied to the search procedure. For example, elements Si and O with the "only"
constraint are used in the periodic table to match all Si-O compounds, and on the Names tab the mineral name "cristobalite"
can be used to find only those compounds that are called by that mineral name (this of course could be used by
itself in the match procedure).
The X-ray Diffraction Lab is located here:
Scott Rudolph Hall, Environmental, Earth, and Planetary Sciences, Room 152
For information about visitor access and parking see: WUSTL Visitor Parking.
On the WUSTL parking map Rudolph Hall is building 105.
Here is the mailing address if you are sending samples:
Paul Carpenter
Rudolph Hall Room 110
Department of Environmental, Earth, and Planetary Sciences, CB1169
Washington University
1 Brookings Drive
St. Louis, MO. 63130-4899
Email (preferred): paulc@wustl.edu
Phone: (314) 935-2585
Return to Top
Return to Analytical Facility Page
Return to EPS Home Page
Document last updated on
by Paul Carpenter